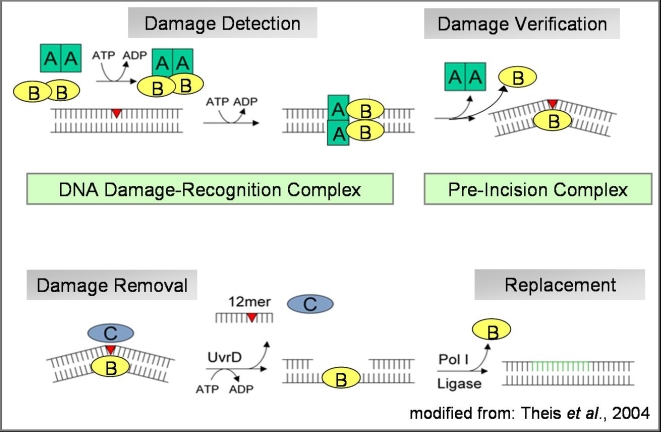
Maintenance of the correct genetic information is crucial for all living organisms. DNA damage, if not repaired,
can cause mutations leading to cancer and may also be involved in aging. Nucleotide Excision Repair (NER) is a
major DNA repair mechanism which is found in all biological live forms.
The repair of DNA by removal of the damaged
DNA fragment through dual incision on both sides of the lesion allows this system to repair a wide variety of
different defects. These include removal of bulky adducts such as pyrimidine dimers induced by UV radiation,
benzo[a]pyrene-guanine adducts caused by smoking, as well as thymine psoralen and guanine-cisplatin adducts caused
by chemotherapeutic drugs.
In humans, dysfunctional NER is responsible for three severe diseases, namely Xeroderma
pigmentosum (XP), Cockaynes syndrome (CS) and trichothiodystrophy (TTD). We study both prokaryotic and eukaryotic NER.
Although the UvrABC endonuclease-mediated excision repair in Escherichia coli is biochemically well
characterized, it is not clear how damaged DNA is recognized and how the components of this system
identify this broad substrate range. We are attempting to address this problem by the structural
characterization of the individual proteins of this system and their complexes both with and without
their substrates, using x-ray crystallography supported by biochemical and biophysical methods.
As a complementary approach we apply atomic force microscopy
(AFM)
in collaboration with the Tessmer lab to obtain images of these complexes under physiological conditions.
We have determined the three-dimensional structure of UvrB as a first step towards understanding
the recognition process of NER. The UvrB structure clearly shows that the enzyme functions as a
helicase adapted to the unique requirements of DNA repair. Based on our crystal structure we propose
how a tight complex between UvrB and DNA can be formed, ensuring differentiation between damaged and
non-damaged DNA. We are in the process of determining structures of UvrB in the presence of damaged
DNA containing different lesions to obtain insight into how these lesions can be specifically recognized.
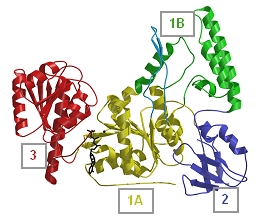
Structure of UvrB
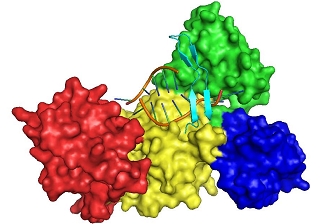
Structure of the pre-incision complex
We have further investigated the NER endonuclease UvrC from Thermotoga maritima. UvrC consists of several domains as shown in the figure below. Although the structures of both the N- and the C-terminal endonuclease domains could already be determined, the NER mechanism remains unclear. To decipher this process, we are striving to obtain the structure of the complex of the NER proteins UvrB and UvrC bound to damaged DNA. For these studies a single amino acid mutant of UvrC is used, which is deficient with respect to the 3' endonuclease activity but structurally similar to the wildtype protein. This mutant allows us to scrutinize the DNA-bound complex before DNA-incision takes place. The structure of the specific UvrBC-DNA complex will allow an enhancement of our understanding of the mechanism of damage verification and subsequent removal.
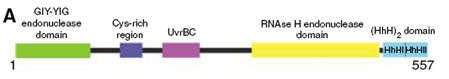
B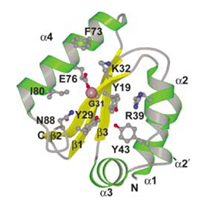
Structure of the N-terminal endonuclease
domain of UvrC
C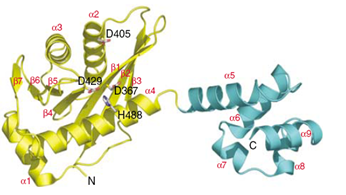
Structure of the 5' endonuclease and (HhH)2
domain of UvrC
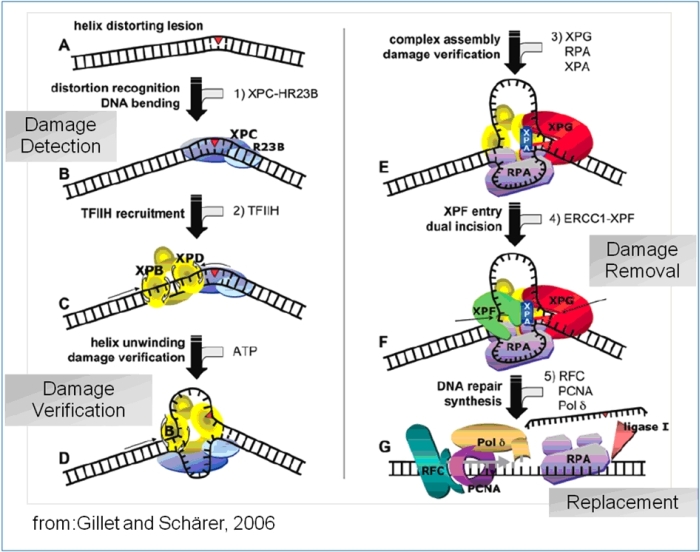
Schematic Overview of the eukaryotic nucleotide excision repair pathway, leading from damage recognition to damage
verification to damage removal and resynthesis
More than 30 proteins are involved in the eukaryotic repair pathway and a general scheme has emerged how
the cascade proceeds through either a “bipartite substrate discrimination” or a “multipartite damage
recognition” model leading to the subsequent steps in the repair pathway. A modification in the DNA backbone leading to the
disruption of base-pairing is recognized by the combined action of XPC and HR23B. Both proteins recruit the 10-subunit transcription
factor TFIIH to this site. XPD and XPB, two helicases in TFIIH, open the DNA around the lesion in an ATP dependant fashion, which is
the first catalytic step in this reaction pathway, leading to a conformational change that allows the recruitment of additional NER
factors.
A second more important function of the two helicases is the step of damage verification. Analogous to the UvrB protein
in prokaryotic NER both XPB and XPD separate the DNA around the lesion. If either of the helicases encounters the lesion it is stalled,
thus verifying the damage and ensuring that the backbone distortion is not due to a specific DNA sequence. This process was
termed “enzymatic proof-reading” and supports the bipartite damage recognition model in which the function of XPC/HR23B
is limited to the observation of a backbone distortion while the two helicases are required to verify the damage through their helicase
activity.
Once this stalled complex is formed RPA, XPA and XPG, the endonuclease responsible for 3' incision, are recruited to form the
pre-incision complex. The ERCC1-XPF endonuclease complex responsible for 5' incision is recruited last; at this point the dual
incision reaction catalyzed by ERCC1-XPF and XPG results in an excised oligomer containing the lesion with a length of 24-32
nucleotides. The repair process is completed by DNA polymerase δ or ε, which both require the presence of PCNA.
The nick between the newly synthesized DNA and the parental strand is closed by DNA ligase I.
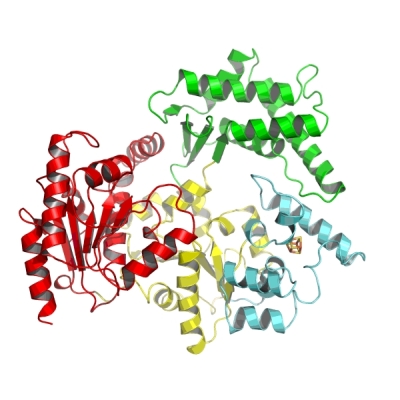
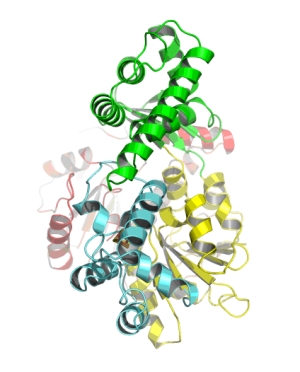
Overall structure of the XPD protein. The two views are rotated 90° relative to each other. The two RecA like helicase domains are shown in yellow and red. The 4Fe4S cluster containing domain is depicted in cyan and the alpha-helical domain, which completes the ring of the “donut” is shown in green.
We recently solved the crystal structure of the nucleotide excision repair protein XPD from Thermoplasma acidophilum,
which shares high sequence identity to human XPD. The XPD protein forms a donut-shaped molecule, which is able to separate two
DNA strands and scan the DNA for damage.
Based on our structural information we were able to deduce possible disease causing mutations thus aiding in the fundamental
understanding how one protein can be involved in three different severe diseases. The figure below shows a space filling
representation of the point mutations leading to the diseases xeroderma pigmentosum (XP), Cockayne syndrome (CS),
and trichothiodystrophy (TTD). The mutations are labeled according to their residue number for T. acidophilum XPD in
black and according to the disease they cause in human XPD, green (XP), blue (TTD) and grey (XP/CS).
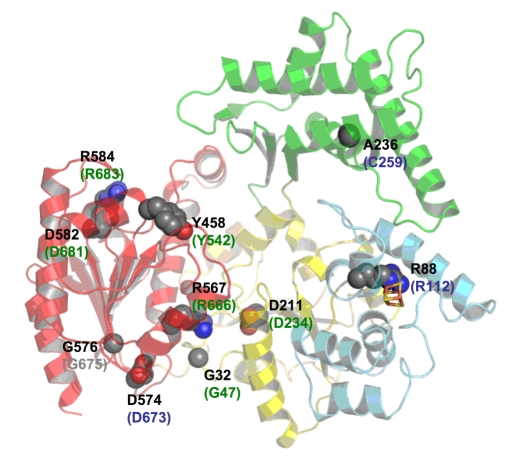
Point mutations leading to xeroderma pigmentosum (XP), Cockayne syndrome (CS) or trichothiodystrophy (TTD) are shown in a space-filling representation and are labelled according to their residue number in T. acidophilum in black and according to the human XPD protein and the disease they cause in humans in green (XP), blue (TTD) and grey (XP/CS)