DNA-dependent DNA polymerases are required for the accurate replication
and repair of DNA. Mitochondrial DNA is replicated and repaired by a
nuclear-encoded DNA polymerase, Polγ, distinct from the polymerases
that replicate and repair nuclear DNA.
Polγ is composed of two
subunits, a catalytic subunit of 125-140 kDa related to the family A of
DNA polymerases, and an accessory subunit of 35-51 kDa . The accessory
subunit, PolγB, has been characterized as a processivity factor for the
polymerase. Upon interaction with the catalytic subunit, PolγB
increases the affinity of the polymerase for DNA and promotes tighter
nucleotide binding, increasing the polymerization rate.
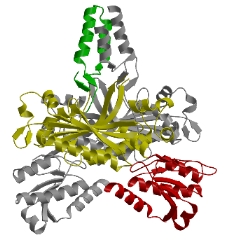
We determined
the crystal structure of mouse PolγB, a core component of the
mitochondrial replication machinery. PolγB shows high similarity to
glycyl-tRNA synthetase (yellow and red domains) and dimerizes through
an unique intermolecular four-helix bundle (green domain). A human
PolγB mutant lacking the four-helix bundle failed to dimerize in
solution or to stimulate the catalytic subunit PolγA, but retained the
ability to bind with PolγA to a primer-template construct, indicating
that the functional holoenzyme contains two PolγB molecules.
The evolutionary relationship of PolγB to aminoacyl-tRNA synthetases (aaRS)
is reflected in conservation of nucleic acid binding properties, since
surface loops involved in tRNA recognition by aaRS appear to be
important for the interaction of PolγB with folded ssDNA. The
processivity factor PolγB is distinct from the sliding clamps like PCNA
and has little structural similarity to thioredoxin or UL42. Other
mutants retained stimulatory activity but lost the ability to bind
folded ssDNA. These results suggest that the PolγB dimer contains
distinct sites for PolγA binding, dimerization, and DNA binding.
Exocyclic DNA adducts, including 3,N4-ethenodeoxycytidine (&epsilon dC),
1,N6-ethenodeoxyadenosine (ε dA) and N2, 3-ethenodeoxyguanosine
(&epsilon dG), are generated by the reaction of metabolites derived from vinyl
chloride and ethyl carbamated with DNA. Exocyclic DNA adducts also form endogenously,
most likely due to lipid peroxidation and oxidative stress, and have been identified
in human DNA.
Patients suffering from Wilson's disease and primary hemochromatosis
accumulate exocyclic adducts in liver DNA. Presumably, DNA is damaged by reactive
oxygen species, the formation of which is promoted by elevated levels of copper or
iron. Both &epsilon dA and &epsilon dC have been used as biomarkers for DNA damage induced
by oxidative stress or lipid peroxidation, processes that may play a role in the etiology
of cancer and chronic diseases associated with aging.
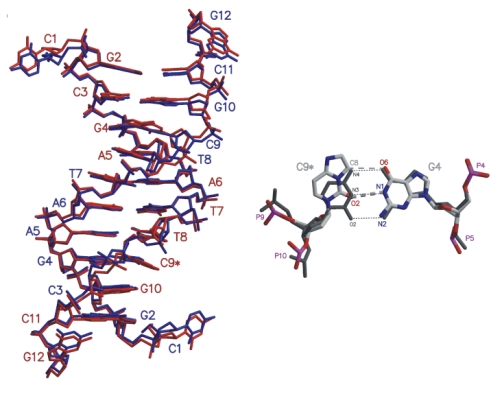
The exocyclic adduct 3,N4-etheno-2'-deoxycytidine (&epsilon dC), positioned
opposite deoxyguanosine was structurally
characterized in the double-stranded dodecanucleotide d(CGCGAATT &epsilon CGCG). Structural
changes within the duplex are limited to the &epsilon C:G and adjacent T:A and G:C base
pairs. The standard Watson-Crick base pairing scheme, retained in the T:A and G:C
base pairs, is blocked by the etheno bridge in the &epsilon C:G pair. In its place,
a hydrogen bond involving 02 of &epsilon C and N1 of G is present. Superposition with
the crystal structure of a DNA duplex containing a T:G wobble pair shows similar structural
changes imposed by both mismatches.
Sulfite oxidase is ubiquitous among animals and catalyzes the
physiologically vital oxidation of sulfite to sulfate, the terminal
reaction in the oxidative degradation of the sulfur containing amino
acids cysteine and methionine. Sulfite oxidase belongs to the
molybdenum cofactor containing family of enzymes which catalyze
important transformations in the global sulfur, nitrogen and carbon
cycles.
In humans, genetic deficiency of sulfite oxidase leads to
severe neurological abnormalities, mental retardation and, in several
cases, attenuated growth of the brain. The structure of chicken liver
sulfite oxidase represents the first step towards understanding the
metabolic disease sulfite oxidase deficiency at the atomic level, but
additional questions arise which we try to address with our current
work. These include structural changes induced by the mutations leading
to sulfite oxidase deficiency in humans; identification of residues
which are involved in electron transfer between the active site
molybdenum and the heme; structural studies on the complex formed
between sulfite oxidase and cytochome c and the intermolecular electron
transfer between these two proteins.
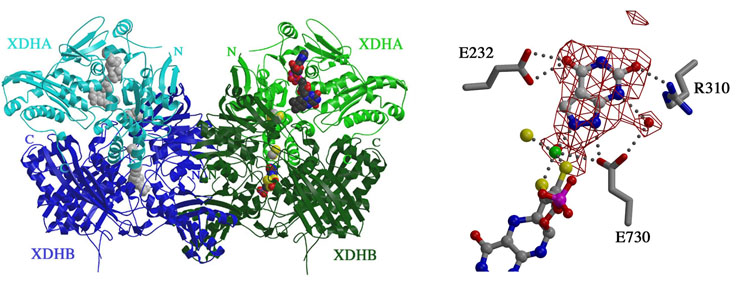
Xanthine Dehydrogenase, a molybdo/iron-sulfur/flavoprotein, catalyzes
the oxidation of hypoxanthine to xanthine and then to uric acid. XDH
from R. capsulatus is composed of two unique subunits denoted XDHA and
XDHB (left panel). XDHA, which contains the flavin adenine dinucleotide
(FAD) and iron/sulfur center domains, forms a dimer with XDHB, the
molybdenum cofactor (moco) harboring subunit. Two of these AB complexes
dimerize via XDHB to form the functional heterotetramer, A2B2.
In the co-crystal structure of XDH inhibited by alloxanthine, the inhibitor is
positioned deeply in the substrate binding pocket (right panel), much
closer to the Mo ion of the moco than the salicylate observed in the
bovine structure. In fact, our crystallographic data supports the
conclusion drawn from biophysical studies that a nitrogen atom of the
inhibitor binds directly to the molybdenum without a bridging oxygen
ligand.